
Nel 1941 il dottor Harry Klinefelter pubblicò il resoconto delle sue ricerche che riguardavano nove uomini con caratteristiche fisiche specifiche: ipotrofia testicolare, ghiandole mammarie sviluppate e diminuzione dei peli corporei. Erano affetti dalla sindrome di Klinefelter o XXY.
IN BREVE
La Sindrome di Klinefelter, non ha nulla a che vedere con la teoria gender ma è una malattia cromosomica maschile non ereditaria caratterizzata dalla presenza di un cromosoma X in più. Rientra tra le patologie aneuploidi (dovute ad un’anomalia nel numero dei cromosomi). A causa di questa condizione i pazienti affetti presentano un patrimonio genetico XXY con 47 cromosomi o, date le ultime scoperte scientifiche, da patrimoni molto più rari XXXY o XXXXY con 48 o 49 cromosomi rispettivamente (a fronte dei normali 46 cromosomi). In queste ultime forme tuttavia il paziente, oltre ad avere la classica sintomatologia associata al Klinefelter, è caratterizzato anche da gravi deficit cognitivi e alterazioni fisiche.

I cromosomi sono lo scrigno dei nostri geni, ovvero delle caratteristiche generali che caratterizzano la specie umana e delle caratteristiche specifiche che distinguono ogni essere umano dall’altro. La specie umana ha 23 coppie di cromosomi (quindi in tutto sono 46) di cui 22 sono dette somatiche (o autosomi) e 1 coppia è il cromosoma sessuale o eterosoma (XX per le femmine, XY per i maschi). Ogni coppia di cromosomi è determinata da un processo biologico che prende il nome di mitosi: ogni cellula madre dà vita a due cellule figlie identiche con corredo cromosomico completo (46). Per i cromosomi sessuali però non è così, altrimenti per ogni riproduzione si avrebbero individui con patrimonio genetico raddoppiato, quadruplicato ecc. La replicazione sessuale, o meiosi (che avviene durante la fase S del ciclo cellulare) non è altro che l’unione di due mitosi senza che vi sia tra esse la duplicazione del DNA. In questo modo durante la meiosi I (detta anche riduzionale), che è preceduta dalla duplicazione del materiale genetico (fase pre-S), da una cellula diploide si generano due cellule figlie con patrimonio genetico dimezzato (aploidi=23 cromosomi) contenenti ognuna due coppie di cromatidi fratelli che formano i cromosomi omologhi. La meiosi II (detta equazionale) è del tutto simile alla mitosi: si conclude quindi il processo mediante il quale da due cellule madri aploidi si generano quattro cellule figlie identiche che non saranno altro che le cellule uovo e gli spermatozoi.
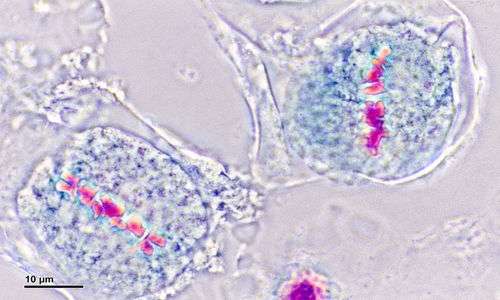
I soggetti affetti da malattie aneuploidi come la sindrome di Klinefelter, sono quelli in cui durante la mitosi o la meiosi si determina un processo anomalo di non-disgiunzione di uno dei due cromatidi che porta, dunque, alla permanenza di uno o più cromosomi:
- se l’anomalia si genera durante le meiosi, il soggetto avrà cellule sessuali con 24 cromosomi
- se invece si genera durante la mitosi, il soggetto avrà 47 cromosomi nel suo patrimonio genetico
In quest’ottica si inserisce un concetto importante che spiega perché alcuni soggetti con sindrome di Klinefelter hanno manifestazioni più gravi e importanti della patologia mentre altri hanno una sintomatologia attenuata: il mosaicismo genetico. Se la non-disgiunzione avviene durante la meiosi, ovociti o gli spermatozoi hanno un patrimonio genetico alterato prima ancora della fecondazione. In questo caso il paziente sarà un 47, XXY. Se la non disgiunzione avviene invece durante la mitosi e, dunque, dopo la fecondazione, il genoma sarà misto e l’errore si manifesta durante la divisione cellulare nell’embrione. In questo caso, alla fine dello sviluppo embrionario, ci saranno alcune cellule con patrimonio XY (46 cromosomi) e altre con patrimonio XXY (47 cromosomi). Questi pazienti avranno una sintomatologia attenuata.
Il testosterone che non c’è
A differenza di quanto si crede, il paziente con sindrome di Klinefelter inizia ad avere i primi sintomi relativi alla sua condizione sin dalla prima infanzia anche se, naturalmente, la sindrome diverrà clinicamente rilevabile con l’arrivo della pubertà quanto i pazienti 47,XXY avranno un iposviluppo dei caratteri sessuali secondari maschili. Il testosterone è importante in tutte le fasi della vita, dall’infanzia all’età adulta.

- durante i primi 6-9 mesi di vita, l’ipotalamo dei pazienti con sindrome di Klinefelter stimola la produzione di gonadotropine (come se fosse in pubertà) determinando quindi un aumento del testosterone circolante. Secondo uno studio condotto al Policlinico Umberto I di Roma, i livelli di testosterone sono deleteri per il corretto sviluppo del cervello del bambino tanto da essere inversamente proporzionali alla capacità di acquisire la parola;
- in età neonatale è possibile, in un numero limitato di casi, avere a che fare con ipo-sviluppo del pene e con criptorchidismo
- durante tutta la fase pre-puberale i bambini hanno una concentrazione ridotta dell’FT3, l’ormone tiroideo libero fondamentale per un corretto sviluppo. In questa fase i pazienti possono mostrare dei sintomi molto poco specifici quali ritardo mentale, dislessia, astenia (riduzione della forza muscolare). Normalmente in questa fase il testosterone predispone alla successiva fase determinando le prime differenze fisiche che vigono tra femmina e maschio
Con la pubertà arrivano le manifestazioni tipiche della sindrome di Klinefelter. In soggetti normali in questa fase il testosterone determina lo sviluppo dei caratteri sessuali maschili quali aumento dei peli, riduzione del tono della voce, aumento della massa muscolare e rafforzamento dello scheletro. Sembra che il testosterone protegga poi i maschi in età adulta da patologie quali l’osteoporosi perché esso viene convertito in estrogeni che favorisce il deposito di calcio nelle ossa.

I pazienti 47,XXY entrano normalmente in pubertà ma, poiché i loro livelli di testosterone non sono nella norma, essa subisce un rallentamento importante e una precoce conclusione. I segni/sintomi caratteristici sono:
- ipogandismo: le gonadotropine, pur essendo prodotte in maniera consona allo sviluppo, non sono in grado di interagire con i recettori posti sulle cellule testicolari e questo oltre a causare un aumento delle stesse nel sangue e una loro conseguente escrezione urinaria, determina un anomalo sviluppo dei testicoli, che risultano più piccoli del normale, e un’alterata produzione di testosterone;
- spermatogenesi incompleta o completamente assente (azoospermia): il testosterone stimola la maturazione dei tubuli seminiferi e la spermatogenesi e dunque nei soggetti affetti da sindrome di Klinefelter queste due processi avvengono in maniera anomala (nei soggetti privi di mosaicismo) o non avvengono affatto (soggetti con mosaicismo).
- ginecomastia: con la riduzione del testosterone libero aumenta la produzione periferica di estrogeni che trovano recettori a livello della ghiandola mammaria determinandone una crescita. Esistono due forme di ginecomastia, quella vera che è dovuta ad una proliferazione non tumorale dei dotti e dello stroma mammario, quella falsa in cui l’aumento delle dimensioni della mammella è successivo ad un eccessivo accumulo di tessuto adiposo.
Il testosterone mancante nei pazienti 47,XXY è dunque la causa del mancato sviluppo dei caratteri sessuali secondari maschili: cambiamento del tono della voce, comparsa dei peli e della barba, spermatogenesi e sviluppo della massa muscolare.
A parte queste alterazioni fisiche, il paziente con sindrome di Klinefelter ha anche problemi di natura psicologica dovuti, in una minoranza dei casi a deficit cognitivi causati dalla malattia, nella maggior parte delle situazioni alla loro condizione in relazione all’ambiente che li circonda: un soggetto con sindrome di Klinefelter è molto insicuro, ansioso, tende alla sudditanza ma, soprattutto, è fortemente predisposto a sviluppare la depressione.
Uno studio scientifico pubblicato nell’agosto di quest’anno ha portato alla luce lo strano caso di un paziente a cui non era mai stata diagnosticata la sindrome di Klinefelter ma che riportava una storia personale costellata di rapporti sessuali casuali e ipersessualità non associata sempre a libido. Dal momento che questo soggetto fu posta diagnosi di 47,XXY questo caso ha gettato nuovi dubbi sulla sindrome di Klinefelter: la riduzione del testosterone è una delle cause principali della riduzione della libido, perché questo paziente ha dimostrato invece un’ipersessualità tanto spiccata? Rispondere a questa domanda è importante soprattutto dal punto di vista terapeutico: trattare con testosterone soggetti con sindrome di Klinefelter associata a desiderio di ipersessualità significherebbe aumentare la libido dei pazienti stessi.
Sindrome di Klinefelter: come diagnosticarla?
I pazienti con sindrome di Klinefelter hanno una diagnosi soprattutto clinica: l’ipogonadismo è, come si è visto, la condizione clinica principale a cui si associano però altre manifestazioni tra cui distribuzione femminile del grasso corporeo, habitus eunucoide, intolleranza al glucosio o diabete mellito e, in alcuni casi, anomalie ossee in particolare la sproporzione tra la lunghezza degli arti superiori e il tronco (questo dipende dal fatto che fisiologicamente il testosterone durante la pubertà impedisce che le ossa crescano troppo).
La diagnosi è avvalorata da esami di laboratorio che evidenziano un aumento delle gonadotropine (soprattutto FSH), una concentrazione del testosterone ai limiti inferiori della norma e un proporzionale aumento degli estrogeni. Il paziente, tuttavia, viene ufficialmente dichiarato affetto dalla sindrome di Klinefelter soltanto dopo la mappatura cromosomica o cariotipo.
L’alimentazione e lo stile di vita che aggravano la malattia
La mancanza di testosterone non ha ripercussioni negative soltanto durante lo sviluppo puberale ma determina un aumento del colesterolo circolante (perché il testosterone origina a partire dal colesterolo e, dunque, una ridotta conversione dell’ormone determina un aumento del colesterolo in circolo) e favorisce l’osteoporosi. Queste condizioni predispongono ovviamente allo sviluppo di sindrome metabolica, ipertensione, diabete mellito e, in alcuni casi, tumore al seno.
Un ulteriore studio ha dimostrato che i soggetti affetti da sindrome di Klinefelter tendono ad avere una zona avascolare della fovea e una retina più sottile rispetto ai soggetti sani. Questo, associato a probabili alterazioni architettoniche e morfologiche dell’occhio dei pazienti 47,XXY, dovrebbe indurre a pensare che essi siano soggetti a problemi di vista anche importanti durante la loro vita. Tali alterazioni sono probabilmente dovute al cromosoma X soprannumerario.
Convivere con la Sindrome di Klinefelter
Partendo dal presupposto che, essendo una malattia genica, oggi non esiste possibilità di guarigione, ai pazienti affetti da sindrome di Klinefelter è destinato un trattamento sostitutivo:
- somministrazione graduale di testosterone a partire dalla pubertà e, in alcuni casi da continuare per tutta la vita, per aumentarne i livelli in circolo;
- trattamento chirurgico per risolvere la ginecomastia, asportando tessuto adiposo e ghiandolare. I pazienti con sindrome di Klinefelter tendono alla depressione sociale perché non sono a loro agio nel proprio corpo;
- ricorrere a procedure quali la fecondazione in vitro o iniezione intracitoplasmatica dello sperma per aggirare il problema della sterilità (quest’ultimo trattamento è però utilizzabile soltanto in pazienti con sindrome di Klinefelter che hanno una quota minima di spermatozoi maturi).
Nonostante quest’approccio terapeutico non permetta al paziente di guarire ha una grandissima valenza dal punto di vista psicologico perché permette al soggetto con sindrome di Klinefelter di riacquistare la sua autostima.
Fonte
- Is there any clinical relevant difference between non mosaic Klinefelter Syndrome patients with or without Androgen Receptor variations?
Nature - A case of Klinefelter syndrome with hypersexual desire.
PubMed - Aberrant ocular architecture and function in patients with Klinefelter syndrome.
PubMed - Klinefelter syndrome presenting as behavioral problems in a young adult
Nature