
“Cos’è un salto di specie? Come fa un virus a trasmettersi da un pipistrello ad un uomo?” probabilmente rientrano tra gli interrogativi che un po’ tutti si sono posti dall’inizio della pandemia da Sars-Cov-2. La storia (e diverse ricerche), però, ci svelano che questo non è un fenomeno recente e di cui potrebbe essere l’uomo stesso la causa.
IN BREVE
Indice
SALTO DI SPECIE: COS’È?
Il “salto di specie” o “zoonosi” (in inglese spillover, parola cercatissima su Google a marzo 2020 a causa dell’innesco della pandemia da Covid-19) rappresenta il fenomeno per cui un agente patogeno riesce a trasmettersi (“a saltare”, appunto) da animali vertebrati all’uomo. Questa è una definizione generale: non sono sempre gli animali, infatti, ad essere la sorgente del contagio. Alcune volte i microrganismi sono trasmessi all’uomo da acqua e cibi contaminati. Tuttavia il salto di specie rappresenta uno dei fattori scatenanti di epidemie/pandemie ed emergenze sanitarie e le aree cosiddette di “confine dell’ecosistema” sembrano essere le maggiori responsabili di questi eventi. Questa ipotesi si basa in gran parte sul sospetto che i tassi di diffusione del salto di specie siano più elevati in paesaggi frammentati e nelle aree in cui l’uomo vive in prossimità della fauna selvatica. Al contrario di come si potrebbe pensare, infatti, questo fenomeno è tutto fuorché di recente acquisizione ed accompagna l’uomo da migliaia di anni. Circa 300 diversi virus, rickettsie, batteri, funghi, protozoi ed elminti sono noti, ad oggi, per la loro capacità di effettuare il salto di specie negli umani.

I casi più (tristemente) famosi di salto di specie dei virus nella storia? Si potrebbe partire col citare quello effettuato dal virus dell’Ebola, causa dell’epidemia sviluppatasi a metà degli anni ’70 nella Repubblica Democratica del Congo caratterizzata da elevata contagiosità e portatrice di una febbre emorragica con tasso di mortalità il 90% dei malati. L’epidemia ha causato la morte di 12 mila persone ed il suo instaurarsi sembra essere collegato ad i contatti esistenti tra l’uomo e la fauna selvatica infetta al confine tra le foreste rurali. Anche l’AIDS (sindrome da immunodeficienza acquisita), esplosa negli anni ’80 deve la sua provenienza alla zoonosi. Il virus HIV dell’immunodeficienza delle scimmie, infatti, ha eseguito il salto di specie trasferendosi all’uomo. Dall’inizio dell’epidemia si sono contati nel mondo 35 milioni di morti e negli ultimi anni la media di decessi che viene registrata è di 800 mila persone all’anno. L’emergenza dovuta all’influenza suina A/H1N1 (infezione virale acuta dell’apparato respiratorio) dalla complessa storia epidemiologica si sviluppò nel 2009 (probabilmente dai suini in Messico) con sintomatologia simile a quella della classica influenza. Essa, tuttavia, causò un numero di vittime tra i 100.000 ed i 400.000 solo nel primo anno, colpendo soprattutto anziani, bambini e persone affette da patologie croniche. La precedente SARS si diffuse in Cina e causò il contagio di 8 mila persone con una mortalità del 10%. Anche in quel caso, i pipistrelli furono il serbatoio naturale del virus ed il passaggio all’uomo avvenne tramite un ospite intermedio, lo zibetto, un piccolo mammifero simile al gatto che viene catturato e venduto nei mercati alimentari. Per quanto riguarda la Mers (Sindrome respiratoria del Medio Oriente), la sua comparsa si è registrata per la prima volta in Arabia Saudita nel 2012. Sono stati i cammelli e i dromedari a fare da ospiti intermedi del virus prima di passare all’uomo. Meno citato ma sicuramente più antico è invece il salto di specie del virus del morbillo il cui spillover va retrodatato di ben 1400 anni. Il morbillo umano è una malattia infettiva provocata dal Measles morbillivirus (MeV), un virus a RNA appartenente alla famiglia dei Paramyxoviridae e strettamente imparentato con quello della peste bovina, ora eradicato: il Rinderpest morbillivirus (RPV). Secondo gli studi più recenti il morbillo potrebbe aver compiuto il suo salto di specie proprio dai bovini e i due virus si sarebbero separati dal loro antenato comune più recente tra l’XI e il XII secolo. Un altro virus ad aver effettuato il salto di specie da equini e roditori all’uomo è invece il virus dell’Epatite E di cui però, fortunatamente, sono stati riscontrati pochi casi.
Quali sono le cause del salto di specie?
Il salto di specie è legato al susseguirsi di tre stadi fondamentali:
- La singola infezione iniziale di un nuovo ospite senza trasmissione successiva (cosiddetta dead-end);
- Spillover che provocano catene locali di trasmissione nella nuova popolazione ospitante prima che l’epidemia svanisca (formazione dei focolai);
- Epidemia o trasmissione endemica sostenuta da un ospite ad un altro (host-to-host) nella nuova popolazione;
Tali steps possono essere riassunti in: esposizione, infezione, diffusione ed adattamento. Variabili che influenzano il “successo” dell’insorgenza di una malattia possono inficiare ciascuno di questi passaggi e comprendono il tipo e l’intensità dei contatti tra l’ospite “serbatoio” (donatore o reservoir) ed il nuovo ospite (ricevente), le barriere che l’organismo oppone all’infezione (ad esempio quelle cellulari), fattori unicamente virali che permettono infezioni più o meno efficienti nel nuovo ospite e la diffusione del virus stesso all’interno della nuova popolazione ospitante.

Dunque, quali sono i fattori reali che portano o ostacolano il salto di specie? La probabilità che avvenga il salto di specie è determinata dalle barriere che rendono limitato il contatto tra il potenziale nuovo ospite e la specie infettante. La barriera primaria che il virus può incontrare è data da quella ecologica. Il contatto tra il donatore ed il ricevente è una condizione preliminare per il salto di specie del virus ed è quindi influenzata dalla separazione geografica, ecologica e comportamentale delle specie donatrici e riceventi. Fattori che influenzano la distribuzione geografica delle specie ospiti (ad esempio, il commercio di animali selvatici e l’introduzione di specie domestiche specie) o che diminuiscono la loro separazione comportamentale (ad esempio, la caccia ad animali selvatici) tendono a promuovere l’emergenza data dalla diffusione della malattia. Le barriere, tuttavia, non esistono solo a livello ecologico. Per infettare un nuovo ospite, un virus deve essere in grado in prima istanza di infettare in modo efficiente le cellule della “vittima”. I processi che possono limitare tale infezione sono molteplici ed includono il mancato ingresso della specie infettante grazie alle naturali barriere biologiche di tipo meccanico e fisico, il mancato od inefficace legame ai recettori cellulari, mancata replicazione del genoma e/o espressione dei geni. Un altro fattore determinante riguarda la specificità del virus per l’ospite. Quando essa viene meno il patogeno deve andare incontro a diversi cambiamenti genetici per poter affrontare le barriere multiple opposte dal ricevente, insomma deve subire diverse mutazioni. Altri ostacoli significativi all’infezione possono includere risposte antivirali innate (come quelle indotte da mediatori quali l’interferone e le citochine) ed altre risposte cellulari che limitano l’infezione da parte di alcuni virus.
Il salto di specie è in aumento a causa dell’uomo?
Ebbene sì, diversi studi affermano che l’uomo, con i suoi cambiamenti a livello di ecosistema, sia “causa del suo male” promuovendo il salto di specie dei virus. In particolare, uno studio condotto dai ricercatori delle università della California e di Melbourne (ancor prima che l’attuale epidemia si diffondesse) ha messo in luce che il tasso di zoonosi è decisamente più elevato quando lo sfruttamento da parte degli esseri umani e la distruzione degli habitat minacciano gli animali selvatici. La ricerca suggerisce, infatti, che tanto maggiore è lo stress provocato dall’uomo nei mammiferi, tanto maggiore è il rischio associato al verificarsi del salto di specie. Tali fattori di stress derivano, ad esempio, dalla cattura di animali selvatici (senza considerare quelli da allevamento, anche loro protagonisti in passato dello spillover), strappati alla loro casa naturale, per immetterli in commercio. Questo stress farebbe aumentare la suscettibilità ad infezioni virali in quanto un maggior numero di infezioni è direttamente proporzionale alla dispersione del virus (tramite, ad esempio, secrezioni). La deforestazione, inoltre, favorisce la scomparsa degli animali meno pericolosi per la nostra specie, lasciando sopravvivere quelli potenzialmente più dannosi. Potrebbe sorgere spontanea una domanda: perché la maggioranza delle zoonosi provengono da pipistrelli, roditori e primati e solo in parte dagli animali domestici? Vi sono un paio di ragioni per cui alcune specie hanno un rischio minore o maggiore di sviluppare virus zoonotici. I mammiferi selvatici con uno stato di conservazione minacciato sono rari, quindi la probabilità che essi incontrino l’uomo è bassa. Possono anche ospitare meno parassiti, dato che le loro popolazioni sono più piccole. Le specie che subiscono o hanno subito una riduzione della popolazione dovuta allo sfruttamento umano – attraverso la caccia, il commercio e l’occupazione- presentano più del doppio di virus zoonotici rispetto a quelli minacciati per altri motivi. La caccia ed il commercio, infatti, rappresentano un modo per gli animali selvatici e gli esseri umani di venire a contatto. L’unica soluzione sembrerebbe essere lo sviluppo di una coscienza in materia, volta a limitare questi fenomeni in modo tale che ogni habitat rimanga inalterato per non andare incontro alle grandi ripercussioni che hanno cambiato il nostro modo di vivere.

QUAL È STATO IL SALTO DI SPECIE ALL’ORIGINE DEL CORONAVIRUS?
I Coronavirus sono virus a RNA a singolo filamento. Secondo il Comitato internazionale per la tassonomia dei virus (ICTV), appartengono alla famiglia Coronaviridae dell’ordine Nidovirales. La sottofamiglia dei coronavirus è composta da 4 generi: Alpha-coronavirus, Beta-coronavirus, Gamma-coronavirus e Delta-coronavirus. Alpha- e Beta-coronavirus sembrano capaci di infettare unicamente i mammiferi. Gamma e Delta-coronavirus infettano gli uccelli ma alcuni di loro possono infettare anche i mammiferi. I primi due generi di coronavirus solitamente provocano sindromi respiratorie nell’uomo e gastroenteriti negli animali. Oltre ai coronavirus altamente patogeni, SARS-CoV, CoV-2 e MERS-CoV, che causano una grave sindrome respiratoria nell’uomo, altri quattro coronavirus (HCoV-NL63, HCoV-229E, HCoV-OC43 e HKU1) possono infettarlo, ma nella grande maggioranza dei casi, inducono solo lievi malattie delle vie respiratorie superiori. Tra le altre patologie causate da Alpha e Beta-coronavirus ve ne sono alcune in specie animali di interesse zootecnico, come il virus della gastroenterite trasmissibile suina, il virus della diarrea epidemica suina (PEDV) ed il coronavirus della sindrome della diarrea acuta suina (SADS-CoV). Tutti i coronavirus che sono in grado di infettare l’uomo sono emersi da eventi zoonotici. SARS-CoV, CoV-2 MERS-CoV, HCoV-NL63 e HCoV-229E hanno probabilmente avuto origine dai pipistrelli mentre HCoV-OC43 e HKU1 dai roditori. Gli animali domestici possono avere ruoli determinanti come ospiti intermedi che permettono la trasmissione del virus dagli ospiti originali naturali all’uomo. Inoltre, gli stessi possono soffrire di malattie causate da coronavirus trasmessi dai pipistrelli. Questo è il caso del PEDV e del SADS-CoV: entrambi rappresentano un recente salto di specie di coronavirus dai pipistrelli ai suini. Poiché 7 delle 11 specie di Alpha-coronavirus e 4 delle 9 specie di Beta-coronavirus sono state identificate solo nei pipistrelli fino ad oggi, si ritiene che i pipistrelli siano il principale serbatoio naturale di Alpha- e Beta-coronavirus.
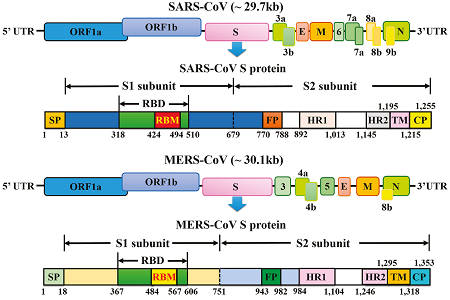
Salto di specie: SARS vs MERS
L’epidemia di COVID-19 (causata dal virus SARS-CoV-2) ha interessanti analogie e differenze con SARS, Sindrome Respiratoria Acuta Grave, e MERS, Sindrome Respiratoria Medio-Orientale, alcune delle quali hanno creato delle vere e proprie pandemie nel corso della storia. Qui si riporta un breve excursus che cerca di fare chiarezza sugli eventi passati in modo che l’attualità sia più facilmente interpretabile.
SARS del 2002
Nel novembre 2002, a Foshan, in Cina, è stato segnalato il primo caso di sindrome respiratoria acuta grave (SARS). Nei mesi successivi, nella Cina continentale sono sorti nuovi casi e nel febbraio 2003 i casi di SARS sono saliti a 300. In seguito, i viaggi hanno diffuso il virus ad Hong Kong e da Hong Kong al Vietnam, al Canada ed a molti altri paesi. Nel marzo 2003, l’OMS si è attivata per costituire una rete di laboratori per isolare l’agente eziologico della SARS. Un notevole sforzo globale ha portato all’identificazione del coronavirus della SARS (SARS-CoV) all’inizio aprile del 2003. L’epidemia si è conclusa a luglio dello stesso anno, dopo essere arrivata in 27 paesi con un totale di 8.096 casi confermati e 774 morti. Altri cinque casi di SARS, derivati da un evento zoonotico strettamente localizzato, sono stati rilevati nel dicembre 2003-gennaio 2004. Da allora, non sono stati rilevati altri casi di SARS. Le misure di prevenzione e di controllo delle infezioni – come la quarantena e l’allontanamento sociale – sono state fondamentali per l’epidemia di SARS. Tuttavia, negli anni successivi, diversi studi hanno evidenziato come i virus legati alla SARS-CoV, potenzialmente in grado di infettare le cellule umane, abbiano continuato a circolare tra i pipistrelli. L’isolamento del SARS-CoV in zibetti, tassi e procioni in un mercato di animali vivi a Shenzen, in Cina, ha fornito la prima indicazione sulla possibile origine di questo nuovo virus. Tuttavia, le analisi successive hanno dimostrato come questi animali siano stati solo ospiti accidentali, perché la presenza di virus simili al SARS-CoV non è mai stata segnalata negli zibetti delle palme o negli animali da allevamento. Solo nel 2005, grazie ad un lavoro di ricerca e di campionamento ambientale, sono stati isolati nei pipistrelli del genere Rhinolophus i virus correlati alla SARS-CoV. Questa scoperta è stata fondamentale per identificare il serbatoio naturale del nuovo coronavirus. In seguito, molti coronavirus correlati alla SARS-CoV (SARSr-CoV) sono stati trovati nei pipistrelli in molte aree della Cina e anche nelle regioni europee, africane e del sud-est asiatico. Questi dati indicano che i SARSr-CoV hanno un’ampia diffusione geografica e che potrebbero essere presenti nei pipistrelli da molto tempo.
MERS del 2012
L’insorgenza nel 2012 della sindrome respiratoria del Medio Oriente (MERS) ha introdotto il secondo coronavirus, altamente patogeno, nella popolazione umana nel XXI secolo. Nel giugno 2012, dieci anni dopo l’epidemia di SARS, un uomo in Arabia Saudita è morto per polmonite acuta ed insufficienza renale. Un nuovo coronavirus, il middle est respiratory syndrome coronavirus (MERS-CoV), è stato isolato dal suo espettorato. In seguito, una serie di casi anomali di grave malattia respiratoria, verificatisi nell’aprile 2012 in un ospedale della Giordania, sono stati diagnosticati retrospettivamente come MERS il quale ha poi continuato ad emergere e a diffondersi nei paesi al di fuori della penisola arabica. Ad oggi, i casi noti di MERS in tutto il mondo sono stati poco meno di 1800 in 27 paesi, con 624 morti. A differenza della SARS-CoV, si ritiene che la MERS-CoV abbia poche possibilità di essere trasmessa direttamente tra gli esseri umani. La maggior parte dei casi noti di MERS è il risultato di salti di specie dai cammelli (o dai dromedari) all’uomo. Sulla base delle conoscenze precedenti sul SARS-CoV, la ricerca del serbatoio naturale di MERS-CoV si è concentrata in prima istanza sui pipistrelli. I test sierologici su campioni di dromedari e cammelli in Oman e nelle Isole Canarie hanno rilevato un’alta prevalenza di anticorpi anti-MERS-CoV in questi animali, suggerendo uno scenario alternativo. Le analisi retrospettive dei tamponi oro-faringei di dromedari e cammelli di un allevamento in Qatar, collegate a 2 casi di MERS umana, hanno permesso di isolare il virus da queste specie in Arabia Saudita e Qatar. Sulla base di queste evidenze, il modello più plausibile per la trasmissione del virus del MERS dagli animali all’uomo è che, all’inizio, il virus si sia diffuso dai pipistrelli – il serbatoio naturale – a dromedari e cammelli il cui ruolo è stato quello di ospiti intermedi. Data l’importanza zootecnica di questi animali in molte aree del mondo, la continua vicinanza di cammelli e dromedari infetti può spiegare i numerosi eventi di salto di specie del MERS-CoV all’uomo.
Sars-Cov-2: l’inizio di una pandemia
I primi casi di COVID-19 sono stati inizialmente collegati al mercato umido (mercato all’aperto dedito alla vendita di beni deperibili) Huanan, a Whuan, in cui, oltre a frutta, verdura, pesce e carne fresca, vengono venduti anche animali vivi. Poiché il salto di specie è indubbiamente alla base della diffusione della Covid-19, una relazione con questa attività non dovrebbe sorprendere. Tuttavia, poiché non tutti i primi casi sono legati al mercato di Huanan, è possibile che la sequenza di eventi che ha portato alla diffusione del SarsCoV-2 all’uomo sia più complicata di quanto inizialmente sospettato. L’analisi di tutti i campioni ambientali ottenuti dal mercato rivela che tutti i virus isolati sono strettamente legati ai primi pazienti di Wuhan. Purtroppo, pare che le autorità cinesi non abbiano eseguito approfonditi campionamenti dalle specie animali presenti nel mercato dunque, è difficile, forse impossibile, fare altre considerazioni. Le prime informazioni sull’origine del SArsCoV-2 sono arrivate dall’analisi del liquido di lavaggio bronco alveolare (BALF) di uno dei sette pazienti affetti da polmonite grave ricoverati nell’unità di terapia intensiva dell’ospedale Jin Yin-Tan di Wuhan. La ricerca nella banca dati per identificare le somiglianze tra il genoma virale isolato ed i genomi virali conosciuti ha indicato che il virus è molto simile al SARSr-CoVs. Ulteriori analisi hanno dimostrato che il virus deriva probabilmente da un coronavirus di pipistrello, in particolare dal coronavirus TG13 che infetta il pipistrello Rinolophus affinis (Bat-CoV-RaTG13). Il lavoro dettagliato di monitoraggio eseguito in Cina negli ultimi anni ha identificato un numero elevato di coronavirus di pipistrelli, tra cui RaTG13. Tuttavia, il ruolo preciso giocato dai pipistrelli nel salto di specie del virus Covid non è chiaro. Curiosamente, i virus CoV-2 più simili sono stati campionati nella provincia dello Yunnan, a 1.500 km da Wuhan. Questo probabilmente significa che, nonostante i grandi sforzi, la nostra conoscenza dei coronavirus dei pipistrelli è ancora largamente incompleta. Inoltre, anche se le somiglianze di sequenza tra RATG13 e CoV-2 (in un range dal 95 al 96%) indicano che questi virus sono strettamente collegati, essi mostrano alcune differenze che probabilmente corrispondono a un’evoluzione avvenuta in più di 20 anni20. Pertanto, un campionamento più accurato identificherà altri virus dei pipistrelli che sono ancora più strettamente legati al CoV-2. Quindi, potrebbero essere necessari diversi anni per identificare con precisione la catena di eventi che hanno permesso al CoV-2 di diffondersi nell’uomo.

Ci si potrebbe chiedere: come possono essere i pipistrelli il veicolo per il salto di specie del coronavirus? Questi animali, pur rappresentando il serbatoio di tali virus, occupano una nicchia ecologica ben separata dagli esseri umani. Pertanto, è altamente possibile che altri mammiferi fungano da ospiti intermedi, in cui il Sars-Cov-2 è stato in grado di acquisire le mutazioni necessarie per diffondersi in modo efficiente tra gli esseri umani. Per identificare questi ospiti intermedi putativi, è imperativo studiare rigorosamente quali tipi di coronovirus possono circolare negli animali in stretto contatto con l’uomo. Infatti, i virus strettamente legati al CoV-2 sono stati trovati in pangolini, che sono illegalmente importati dalla provincia di Guandong e venduti nei mercati cinesi. L’analisi metagenomica dei polmoni da due pangolini, morti in un lasso di tempo compatibile con l’epidemia da COVID-19, ha rivelato che essi sono stati infettati da un virus (dopo definito Pangolin-CoV) strettamente legato al CoV-2 ed altri pipistrelli SARSr-CoV2. I genomi di CoV-2 e Bat-CoV-RaTG13 sono sostanzialmente identici al genoma di PangolinCoV. Pangolin-CoV e CoV-2 condividono, infatti, nella sequenza della proteina spike cinque amminoacidi chiave per entrare nelle cellule ospiti. Questo rende il gene spike del CoV-2 più simile al gene corrispondente (omologo) nel Pangolin-CoV che nel Bat-CoV-RaTG13, suggerendo che le mutazioni possono essere avvenute nel vrus del pangolino e che i pangolini rappresentavano gli ospiti intermedi. Tuttavia, l’origine pangolina del CoV-2 è ancora dibattuta. Infatti, poiché i coronavirus sono caratterizzati da tassi di mutazione (relativamente) bassi, rispetto ad altri virus a RNA, e portano nei loro genomi evidenti segni di ricombinazione, un processo che mescola il materiale genetico, le analisi filogenetiche per determinare le relazioni precise tra i coronovirus – incluso il CoV-2 – sono molto complesse e spesso non definitive. Inoltre, mentre la nostra esperienza passata con i coronavirus suggerisce che l’evoluzione in ospiti intermedi è necessaria per infettare gli esseri umani, non si può escludere che il virus abbia acquisito alcune delle sue proprietà chiave durante un periodo di “fusione criptica” negli esseri umani.
Il salto di specie nei visoni
Da giugno 2020, le autorità danesi hanno segnalato una vasta diffusione di SARS-CoV-2 negli allevamenti di visoni in Danimarca. Il 5 novembre, le autorità sanitarie pubbliche danesi hanno segnalato il rilevamento di una variante SARS-CoV-2 associata al visone con una combinazione di mutazioni non osservate in precedenza (denominate “Cluster 5”) in 12 casi umani nello Jutland settentrionale, rilevate da agosto a settembre 2020. Ad oggi, lo Statens Serum Institut (SSI) in Danimarca ha identificato sette mutazioni uniche nella proteina spike del SARS-CoV-2 tra le varianti co-circolanti nel visone e nell’uomo. L’SSI ha coltivato la variante “Cluster 5” con quattro modifiche dell’amminoacido nella proteina spike, che è stata identificata nel visone e isolata dai 12 casi umani riportati nello Jutland settentrionale. I risultati preliminari hanno suggerito che una minore capacità degli anticorpi di neutralizzare il ceppo del Cluster 5. Tuttavia, ulteriori ricerche hanno suggerito che si tratti di affermazioni di carattere speculativo e che la Cluster 5 possa essere una variante dead-end negli esseri umani per la sua limitata diffusione (e che “ancora” non sia né particolarmente pericolosa né in grado di compromettere il vaccino). Molti di coloro che sono stati infettati lavoravano negli allevamenti e probabilmente sono stati esposti ad una dose virale elevata. Stando ai report dell’OMS, infatti, dalla settimana del 6 giugno 2020 (settimana 24) alla settimana del 16 novembre 2020 (settimana 47), 10386 campioni positivi per la COVID-19 da individui unici sono stati sottoposti a sequenziamento dell’intero genoma (il 17,6% di tutti i campioni positivi nel corrispondente periodo di tempo) Di questi campioni sequenziati, 750 erano varianti di virus associate a visoni infetti. Inoltre, almeno due nuove varianti di SARS-CoV-2 sono state recentemente rilevate nella Danimarca meridionale che non erano geneticamente correlate al ceppo variante originale associato al visone danese. Ad oggi, otto paesi (Danimarca, Lituania, Paesi Bassi, Spagna, Svezia, Italia e Grecia e Stati Uniti d’America, hanno segnalato la presenza della COVID-19 nei visoni d’allevamento all’Organizzazione mondiale per la salute animale (OIE). Il 4 novembre 2020 la Danimarca ha deciso di abbattere tutti i visoni d’allevamento per prevenire la diffusione di tali varianti.

QUALE SALTO DI SPECIE CI ATTENDE?
È possibile predire un nuovo salto di specie? Notevoli progressi sono stati compiuti nell’identificazione dei molteplici fattori che controllano o influenzano il salto di specie dei virus. Ad esempio, consorzi mondiali come il programma USAID PREDICT così come molti laboratori accademici indipendenti in tutto il mondo hanno utilizzato una combinazione di screening tramite PCR (tecnica usata per il sequenziamento genico) per caratterizzare migliaia di nuove sequenze virali nei campioni animali ma arrivare a parlare di predizioni, purtroppo, è prematuro. Naturalmente lo studio sempre più approfondito e la realizzazione di nuove tecnologie (incluso il monitoraggio tramite i modelli epidemiologici) permetteranno di chiarire i meccanismi che consentono l’ingresso, la replica e la trasmissione dei virus che effettuano il salto di specie in modo da rivelare anche gli eventi evolutivi chiave che si sono verificati nelle pandemie precedenti. Le sfide chiave della ricerca attualmente includono i metodi per distinguere i casi infettati direttamente dal salto di specie degli animali (casi primari) ed i casi secondari risultanti dalla trasmissione da uomo a uomo ed i metodi per stimare la trasmissibilità, che sono spesso difficili da ottenere all’inizio di un’epidemia. Chiarendo la serie di steps necessari per passare dall’esposizione alla diffusione epidemica/pandemica, i nuovi modelli riusciranno ad identificare più punti in cui gli interventi strategici potrebbero permetterci di prevenire la diffusione di nuovi virus. L’emergere di nuove malattie virali da animale a uomo è stato, e probabilmente continuerà ad essere, una delle fonti principali di nuove malattie infettive umane per cui solo la reale conoscenza del nemico ci aiuterà a combatterlo.

Fonte
- COVID19: an announced pandemic
Cell Death & Disease - Pathways to zoonotic spillover
Nature reviews microbiology - Cross-species pathogen spillover across ecosystem boundaries: mechanisms and theory
Philosophical transactions of the royal society B - Cross-Species Virus Transmission and the Emergence of New Epidemic Diseases
MICROBIOLOGY AND MOLECULAR BIOLOGY REVIEWS